Cientistas do National Eye Institute (NEI), dos Estados Unidos, usaram células-tronco de pacientes e bioimpressão 3D para criar tecido ocular que deve ajudar a compreender os mecanismos de doenças que causam cegueira. Dentre elas, a Degeneração Macular Relacionada à Idade (DMRI).
A equipe imprimiu uma combinação de células que formam a barreira externa sangue-retina. De acordo com o estudo, publicado na Nature, essa técnica fornece um suprimento teoricamente ilimitado de tecidos derivados do paciente.
Em seguida, entenda como ocorreu a pesquisa e como os resultados podem levar a muitos usos potenciais em aplicações translacionais, incluindo o desenvolvimento terapêutico.
A pesquisa
Segundo os pesquisadores, a Degeneração Macular Relacionada à Idade (DMRI) se desenvolve inicialmente na barreira sangue-retina externa. No entanto, os mecanismos de iniciação e progressão da doença tanto na forma seca como úmida permanecem pouco compreendidos devido à falta de modelos humanizados fisiologicamente relevantes.
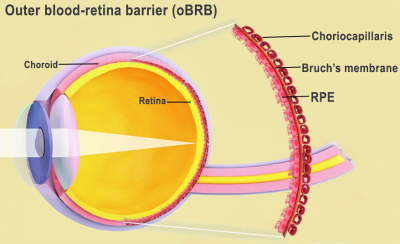
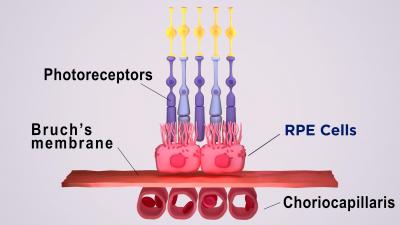
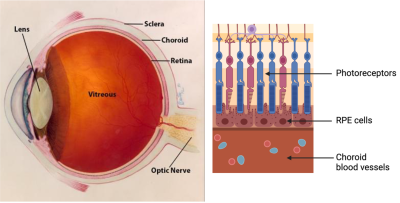
A barreira sangue-retina externa consiste no epitélio pigmentar da retina (RPE), separado pela membrana de Bruch dos coriocapilares ricos em vasos sanguíneos. A membrana de Bruch regula a troca de nutrientes e resíduos entre os coriocapilares e o RPE.
A barreira sangue-retina externa é a interface da retina e da coróide, incluindo a membrana de Bruch e os coriocapilares. Crédito da imagem: National Eye Institute.
Na DMRI, depósitos de lipoproteínas, chamados drusas, se formam na porção externa da membrana de Bruch, impedindo sua função. Com o tempo, o RPE se rompe localmente, levando à degeneração dos fotorreceptores e à perda da visão.
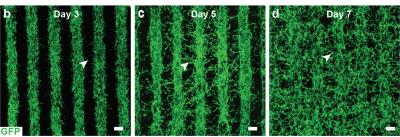
Os cientistas combinaram três tipos de células coroideanas imaturas em um hidrogel: pericitos e células endoteliais, que são componentes-chave dos capilares; e fibroblastos, que dão estrutura aos tecidos. Então, imprimiram o gel em um esqueleto biodegradável e, em poucos dias, as células começaram a amadurecer em uma densa rede capilar.
Os resultados
No nono dia, o time semeou células epiteliais de pigmento da retina no outro lado do esqueleto. O tecido impresso atingiu a maturidade total no dia 42. Análises de tecido e testes genéticos e funcionais mostraram que o tecido impresso parecia e se comportava de maneira semelhante à barreira hematorretiniana externa nativa.
Sob estresse induzido, o tecido impresso exibiu padrões de DMRI precoce, como depósitos de drusas sob o RPE e progressão para DMRI em estágio seco tardio, onde foi observada a degradação do tecido.
A barreira sangue-retina externa do olho compreende o epitélio pigmentar da retina, a membrana de Bruch e os coriocapilares. Crédito da imagem: National Eye Institute.
Também perceberam a aparência de DMRI úmida induzida por baixo oxigênio, com hiperproliferação de vasos coróides, que migraram para a zona sub-RPE. Os medicamentos anti-VEGF, usados para tratar a DMRI, suprimiram o supercrescimento e a migração desse vaso e restauraram a morfologia do tecido.
Os cientistas observaram que a presença de células RPE induz mudanças na expressão gênica em fibroblastos que contribuem para a formação de Membrana de Bruch – algo que foi sugerido há muitos anos, mas não tinha sido comprovado até o modelo desenvolvido por essa pesquisa.
Próximos passos
Crescimento de vasos sanguíneos através de linhas impressas de uma mistura de células endoteliais-pericito-fibroblásticas. No dia 7, os vasos sanguíneos preenchem o espaço entre as fileiras, formando uma rede de capilares. Crédito da imagem: Kapil Bharti.
A equipe enfrentou alguns desafios técnicos, como a geração de um esqueleto biodegradável adequado e a obtenção de um padrão de impressão consistente por meio do desenvolvimento de um hidrogel sensível à temperatura, que deveria alcançar linhas distintas quando frio e se dissolver quando o gel esquentava.
A boa consistência das fileiras permitiu um sistema mais preciso de quantificação das estruturas dos tecidos. Eles também otimizaram a proporção de mistura celular de pericitos, células endoteliais e fibroblastos.
Todo o trabalho resultou em modelos de tecido de retina com muitos usos potenciais em aplicações translacionais, incluindo o desenvolvimento terapêutico. Agora, os cientistas seguem utilizando modelos de barreira sangue-retina impressos para estudar a DMRI.
Além disso, também estão experimentando acrescentar tipos de células adicionais ao processo de impressão, como células imunológicas para modelar melhor o tecido nativo.
Revisado por Paulo Schor, médico oftalmologista, professor livre docente e diretor de inovação da Universidade Federal de São Paulo (Unifesp) e colaborador da Faculdade de Medicina do Hospital Albert Einstein.
Acompanhe o blog da Phelcom e fique por dentro das principais novidades sobre bioimpressão 3D para criar tecido ocular.
Como já sabemos, pacientes com olho seco são mais propensos a sofrerem lesões nas córneas. Agora, pesquisadores da Universidade de Washington, nos Estados Unidos, descobriram que proteínas produzidas por células-tronco que regeneram a córnea podem ser novos alvos para tratar e prevenir essas lesões.
O trabalho foi publicado recentemente no site da revista Proceedings of the National Academy of Sciences.
Em seguida, entenda como foi feito o estudo com camundongos e como os resultados oferecem um novo foco promissor para tratar e possivelmente até prevenir lesões na córnea derivadas do olho seco.
A pesquisa
Pesquisadores rastrearam os movimentos das células-tronco em um olho de camundongo por meio de luz verde fluorescente.Imagem: Universidade de Washington.
Os pesquisadores analisaram genes expressos pela córnea em vários modelos de camundongos – não apenas com olho seco, mas também com diabetes e outras condições.
Eles descobriram que a córnea ativava a expressão do gene SPARCem roedores apenas com olho seco. Além disso, também identificaram que a expressão mais alta desse gene foi associada a uma melhor cicatrização.
Dessa forma, os cientistas acreditam que alguns genes, particularmente o SPARC, podem fornecer alvos terapêuticos potenciais para o tratamento de olho seco e lesões na córnea.
Caso as proteínas encontradas não funcionarem como terapias para ativar essas células em pessoas com síndrome do olho seco no futuro, a equipe levantou a possibilidade de até mesmo transplantar células-tronco límbicas projetadas para prevenir lesões na córnea nestes pacientes.
O pesquisador Rajendra S. Apte explica que milhões de pessoas em todo o mundo, sendo cerca de 15 milhões apenas nos Estados Unidos, sofrem com dores oculares e visão turva como resultado de complicações e lesões associadas à doença do olho seco. “Ao direcionar essas proteínas, podemos ser capazes de tratar com mais sucesso ou mesmo prevenir essas lesões”, ressaltou em comunicado.
Revisado por Paulo Schor, médico oftalmologista, professor livre docente e diretor de inovação da Universidade Federal de São Paulo (Unifesp) e colaborador da Faculdade de Medicina do Hospital Albert Einstein.
Acompanhe o blog da Phelcom e fique por dentro das principais novidades sobre olho seco.
Atualmente, a principal causa de cegueira em adultos americanos é a retinopatia diabética. No entanto, a fonte desse dano parece estar na barriga, segundo um estudo publicado na revista Circulation Research.
Pesquisadores da Universidade do Alabama (EUA) avaliaram o sangue de pacientes com diabetes tipo 1 e um modelo de camundongo com a mesma doença para explorar os mecanismos subjacentes à retinopatia diabética. Os resultados mostram uma maneira de possivelmente prevenir, ou mesmo reverter, o dano ocular.
Em seguida, veja como o estudo ocorreu e como os resultados podem demonstrar, pela primeira vez, que a ruptura da barreira intestinal pode ser implicada na patogênese da retinopatia diabética.
Diabetes tipo 1 e RAS
Primeiro, sabe-se que o diabetes tipo 1 desregula o sistema renina-angiotensina sistêmico (RAS), responsável por regular a pressão sanguínea e outras alterações metabólicas.
Além do RAS sistêmico, existem também redes RAS locais que atuam em diversos tecidos. Uma enzima RAS chave é a ACE2, ou enzima conversora de angiotensina 2. A perda de ACE2 no diabetes ativa o eixo RAS vasodelétrico e diminui o eixo RAS vasoprotetor.
Curiosamente, em um modelo de camundongo com diabetes tipo 1, alimentá-lo com uma cepa bacteriana intestinal modificada de Lactobacillus paracasei, que foi projetada para produzir ACE2 humano, o protege contra a progressão da retinopatia diabética.
Finalmente, sabe-se que a falta de ACE2 no intestino aumenta a permeabilidade intestinal e a inflamação sistêmica.
A pesquisa
Os estudos em humanos compararam pacientes com diabetes tipo 1 em relação às pessoas sem a doença. Os voluntários com o problema foram ainda divididos em três grupos: sem retinopatia diabética, com retinopatia diabética não proliferativa e com retinopatia diabética proliferativa.
Medindo os níveis de certas células imunológicas e biomarcadores no sangue, incluindo antígenos microbianos intestinais, os pesquisadores descobriram que seres humanos com retinopatia tinham um RAS sistêmico desregulado e profundos defeitos de permeabilidade intestinal, que ativavam componentes da resposta imune adaptativa e inata.
Além disso, verificou-se que os aumentos na gravidade da doença se correlacionam com níveis aumentados de biomarcadores de permeabilidade intestinal e um antígeno microbiano intestinal.
Testes em camundongos
Usando o modelo de diabetes tipo 1 do camundongo Akita, os cientistas administraram primeiro o Lactobacillus paracasei produtor de ACE2 aos camundongos, começando no início do diabetes.
Este tratamento probiótico evitou a perda de ACE2 epitelial intestinal normalmente observada em camundongos Akita e, mais importante, evitou danos ao epitélio intestinal e à barreira endotelial. Também reduziu os níveis elevados de açúcar no sangue, conhecidos como hiperglicemia.
Quando o tratamento oral com Lactobacillus paracasei produtor de ACE2 foi suspenso até seis meses após o estabelecimento do diabetes, essa terapia atrasada reverteu a disfunção da barreira intestinal e a retinopatia diabética que já havia se formado nos camundongos, incluindo a redução do número de capilares danificados na retina.
A equipe também encontrou evidências de vários mecanismos que contribuíram para o dano da barreira intestinal reduzido pelo ACE2 e pela redução do açúcar no sangue pelo ACE2.
Para validar os resultados do modelo Lactobacillus paracasei produtor de Akita/ACE2, eles criaram um segundo modelo – uma cepa Akita geneticamente modificada que superexpressa o ACE2 humano nas células epiteliais do intestino delgado.
Os resultados
O trabalho demonstra que o RAS intestinal desregulado resulta na translocação de antígenos microbianos intestinais para o plasma. Esses peptídeos bacterianos ativam o endotélio por meio de receptores toll-like, criando um endotélio inflamatório que tem sido fortemente implicado na patogênese de doenças vasculares, incluindo a retinopatia diabética.
A pesquisa constatou a perda da função da barreira intestinal em seres humanos com diabetes tipo 1 usando biomarcadores da barreira intestinal. Esse aumento na permeabilidade foi associado à ativação de células imunes derivadas do intestino.
“Até onde sabemos, este estudo representa a primeira vez que a ruptura da barreira intestinal foi implicada na patogênese da retinopatia diabética e também relaciona diretamente o vazamento intestinal com a gravidade da retinopatia em seres humanos com diabetes tipo 1”, disse em comunicado a pesquisadora Maria Grant.
Acompanhe o blog da Phelcom e fique por dentro das principais novidades sobre retinopatia diabética.
Há décadas, pesquisadores estudam o uso da terapia genética para cegueira e tratamento de outras doenças oftalmológicas. Por exemplo, um trabalho da Universidade da Pensilvânia (EUA) desenvolveu um tratamento genético experimental que busca recuperar a visão de portadores de amaurose congênita de Leber (LCA) e de deficiência visual grave.
Já um estudo da University College London (UK), de Londres, conseguiu restaurar parcialmente a função dos cones (uma das células da retina) de dois jovens que nasceram com acromatopsia por meio da terapia genética.
Agora, uma nova pesquisa empregou nanotecnologia para criar uma nova abordagem à terapia genética que pode melhorar a forma como os médicos tratam formas hereditárias de cegueira.
O trabalho é realizado pela Oregon Health & Science University e pela Oregon State University, dos Estados Unidos. Os primeiros resultados foram publicados recentemente na Science Advances.
Em seguida, saiba mais sobre os achados e como podem auxiliar no desenvolvimento de novas terapias genéticas baseadas em nanopartículas lipídicas.
A pesquisa
Os cientistas desenvolveram uma abordagem que usa nanopartículas lipídicas (minúsculas bolas de gordura feitas em laboratório) para fornecer cadeias de ácido ribonucléico mensageiro, ou mRNA, dentro do olho. Para tratar a cegueira, o mRNA será projetado para criar proteínas que editam mutações genéticas que prejudicam a visão.
No estudo publicado, a equipe demonstra como o sistema de entrega de nanopartículas lipídicas atinge as células fotorreceptores, tanto em camundongos quanto em primatas não humanos. As nanopartículas são revestidas com um peptídeo, que a equipe identificou como sendo atraído por fotorreceptores.
Como primeira prova de conceito, mRNA com instruções para produzir proteína fluorescente verde foi colocado dentro de nanopartículas.
Os resultados
Depois de injetar esse modelo de terapia genética, a equipe usou uma variedade de técnicas de imagem para examinar os olhos tratados.
O tecido retiniano dos animais brilhou em verde, ilustrando que o invólucro de nanopartículas lipídicas alcançou os fotorreceptores e que o mRNA que ele entregou entrou com sucesso na retina e criou uma proteína verde fluorescente.
Esta pesquisa marca a primeira vez que as nanopartículas lipídicas são conhecidas por terem alvo fotorreceptores em um primata não humano.
Terapia genética para cegueira com AVV
A coautora do estudo, Renee Ryals, explica que mais de 250 mutações genéticas foram associadas a doenças retinianas hereditárias, mas apenas uma tem uma terapia genética aprovada nos Estados Unidos.
Esse tratamento depende amplamente do vírus adeno-associado (AAV) para fornecer moléculas de revisão de genes. Porém, o AVV tem algumas limitações.
O vírus é relativamente pequeno e não pode conter fisicamente máquinas de edição de genes para algumas mutações complexas. E a terapia genética baseada em AAV só pode fornecer DNA, o que resulta na criação contínua de moléculas de edição de genes que podem levar a edições genéticas não intencionais.
Já as nanopartículas lipídicas são uma alternativa promissora porque não possuem restrições de tamanho como o AAV. Além disso, podem fornecer mRNA, que apenas mantém o maquinário de edição de genes ativo por um curto período de tempo, o que pode impedir edições fora do alvo.
O potencial das nanopartículas lipídicas foi ainda comprovado pelo sucesso das vacinas COVID-19 baseadas mRNA, que também foram as primeiras a serem autorizadas nos Estados Unidos, graças à velocidade e ao volume em que puderam ser fabricadas.
“Melhorar as tecnologias usadas para terapia genética pode fornecer mais opções de tratamento para prevenir a cegueira. As descobertas do nosso estudo mostram que as nanopartículas lipídicas podem nos ajudar a fazer exatamente isso”, afirma Ryals.
Próximos passos
Agora, os cientistas estão trabalhando em pesquisas de acompanhamento para quantificar quanto da proteína verde fluorescente é expressa em modelos animais de retina. Além disso, estão desenvolvendo uma terapia com mRNA que carrega o código para moléculas de edição de genes.
Acompanhe o blog da Phelcom e fique por dentro das principais novidades sobre terapia genética para cegueira.
Uma equipe internacional liderada por cientistas da Harvard Medical School e Boston Children’s Hospital, dos Estados Unidos, descobriu uma nova mutação genética que pode ser a causa raiz de quadros graves de glaucoma infantil.
Por meio de tecnologia avançada de sequenciamento do genoma, os pesquisadores identificaram uma mutação no gene da trombospondina-1 (THBS1) em três famílias etnicamente e geograficamente diversas com histórico da doença na infância.
Os novos achados, publicados no Journal of Clinical Investigation, podem levar a uma melhor triagem e tratamentos mais precoces e direcionados para prevenir a perda de visão em crianças com a mutação.
Em seguida, entenda como foi feita a pesquisa.
Glaucoma infantil
O glaucoma infantil, ou congênito, é uma doença rara, mas grave, que se manifesta em crianças desde o nascimento até os 3 anos de idade. Apesar de sua raridade, é responsável por 5% dos casos de cegueira infantil em todo o mundo.
Crianças com o problema geralmente requerem cirurgias já nas primeiras semanas ou meses de vida, eventualmente seguidas por outras operações durante a infância.
Geralmente, há um forte componente hereditário, com vários membros de uma família afetados pela doença. Desse modo, ao entender melhor os genes envolvidos, os testes genéticos podem dar tranquilidade às famílias afetadas para saber se seu filho pode estar em risco de desenvolver a doença.
A pesquisa
Os cientistas usaram um conjunto de dados de mais de 34 mil adultos com glaucoma para identificar 127 genes associados à doença.
Para estudar melhor as mutações genéticas no glaucoma infantil, primeiro foram sequenciadas todas as regiões codificadoras de proteínas dos genes no genoma (exoma) de uma família americana de ascendência europeia-caucasiana que havia feito parte de um projeto de pesquisa anterior.
Os cientistas encontraram uma variante nova e surpreendente na trombospondina -1, uma proteína bem conhecida envolvida em vários processos biológicos importantes, como a formação de novos vasos sanguíneos (angiogênese) e tecidos.
Este gene mutante não foi encontrado em pessoas sem glaucoma infantil, nem em grandes bancos de dados genéticos populacionais. O aminoácido alterado pela mutação foi conservado evolutivamente, indicando um papel importante na função da proteína.
Esse achado levou a equipe a buscar apoio da Flinders University, na Austrália, para ver se havia alguma família com glaucoma infantil com mutações de trombospondina no país. E descobriram que sim.
Para avançar ainda mais essa hipótese, os pesquisadores desenvolveram um modelo de camundongo com a mutação THBS1 e observaram que o animal também apresentava características de glaucoma.
Os resultados
A princípio, presumiu-se que as mutações THBS1 estavam interrompendo a formação de vasos sanguíneos no olho. Entretanto, os modelos animais mostraram angiogênese normal.
Em seguida, a equipe notou que a mutação causava o acúmulo de proteínas anormais da trombospondina nas estruturas de drenagem intraocular do olho, envolvidas na regulação da PIO; o que, por sua vez, levava a elevação da pressão e dano ao nervo óptico e camada de fibras nervosas da retina, provocando perda de visão.
Esta foi a primeira vez que identificaram esse tipo de mecanismo da doença que causa o glaucoma infantil.
Próximos passos
O estudo tem implicações clínicas significativas. Embora ainda haja mais trabalho antes que testes genéticos abrangentes possam ser oferecidos, cada gene encontrado apresenta outra oportunidade para identificar mutações causadoras nessas famílias por meio de triagem.
Terapeuticamente, o conhecimento dessa mutação genética pode levar a tratamentos mais precoces com terapias convencionais. Por exemplo, se um bebê nasce com essa mutação, o oftalmologista pode informar melhor os pais sobre os riscos e desenvolver um plano de tratamento e monitoramento da doença apropriado.
A identificação desse novo mecanismo e gene na raiz do glaucoma infantil também pode levar a novas terapias que visam o combate ao acúmulo de proteínas anormais.
Os pesquisadores também pretendem determinar se outras mutações THBS1 estão envolvidas na doença de início adulto, como glaucoma primário de ângulo aberto ou formas mais leves da doença.
A equipe também continuará procurando novos genes associados ao glaucoma infantil na esperança de um dia desenvolver uma triagem muito abrangente.
Revisado por Paulo Schor, médico oftalmologista, professor livre docente e diretor de inovação da Universidade Federal de São Paulo (Unifesp) e colaborador da Faculdade de Medicina do Hospital Albert Einstein.
Acompanhe o blog da Phelcom e fique por dentro das principais novidades sobre glaucoma infantil.
Uma nova pesquisa do National Eye Institute (NEI), dos Estados Unidos, mostrou, pela primeira vez, como as células em diferentes camadas de tecido do olho são afetadas em pessoas com coroideremia, uma rara doença genética que leva à cegueira.
Para isso, os pesquisadores combinaram técnicas tradicionais de imagem ocular com óptica adaptativa (tecnologia que melhora a resolução da imagem). O estudo foi publicado recentemente na Communications Biology.
Em seguida, saiba mais sobre o trabalho e como os resultados podem trazer avanços no entendimento e posterior tratamento dessa doença e outras relacionadas.
A pesquisa
Os pesquisadores combinaram óptica adaptativa com realce pelo corante indocianina verde para observar células vivas na retina, incluindo fotorreceptores sensíveis à luz, epitélio pigmentar da retina (RPE) e vasos sanguíneos da coróide.
A equipe conseguiu ver em detalhes até que ponto a coroideremia perturba esses tecidos, fornecendo informações que podem ajudar a projetar tratamentos eficazes para esta e outras doenças.
Os resultados
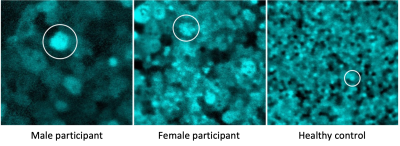
Imagens da retina mostrando células RPE aumentadas (circuladas) em participantes masculinos (esquerda) e femininos (centro) do estudo com coroideremia em comparação com um controle saudável (direita). Crédito: Johnny Tam/National Eye Institute.
A coroideremia afeta mais os homens do que as mulheres porque o gene responsável pela doença está localizado no cromossomo X.
Como os homens têm apenas uma cópia do cromossomo X, uma mutação no gene faz com que os homens desenvolvam sintomas mais graves, enquanto as mulheres – que têm duas cópias do cromossomo X – geralmente apresentam sintomas mais leves, tendo uma cópia ativa do gene no outro cromossomo X.
Uma das principais descobertas do estudo foi que as células RPE estão maiores e disfuncionais em até cinco vezes em homens e mulheres com coroideremia.
As participantes do sexo feminino mostraram uma mistura de células RPE maiores e de aparência mais saudável. Dessa forma, isso pode explicar por que mulheres com a doença apresentam sintomas mais leves.
As camadas de fotorreceptores e vasos sanguíneos foram menos afetadas nos participantes masculinos e femininos do estudo, sugerindo que a interrupção do EPR desempenha um papel importante na coroideremia.
Óptica adaptativa
Células do EPR (ver exemplos circulados) em um participante do sexo masculino com coroideremia, mostrando que células do EPR aumentadas podem ser detectadas usando a abordagem de imagem multimodal de Tam. Crédito: Johnny Tam/National Eye Institute.
A óptica adaptativa utilizada pelos pesquisadores não faz parte dos testes de diagnóstico de rotina em clínicas oftalmológicas.
A equipe descobriu que células RPE maiores podem ser detectadas mesmo quando se usa apenas um oftalmoscópio a laser de varredura disponível comercialmente junto com o corante indocianina verde.
“Usando uma ferramenta existente na clínica, podemos monitorar e rastrear o estado celular da camada RPE. Isso pode ser valioso para identificar quais pacientes se beneficiariam mais com as intervenções terapêuticas”, disse o pesquisador responsável pelo estudo, Johnny Tam, em comunicado.
Revisado por Paulo Schor, médico oftalmologista, professor livre docente e diretor de inovação da Universidade Federal de São Paulo (Unifesp) e colaborador da Faculdade de Medicina do Hospital Albert Einstein.
Acompanhe o blog da Phelcom e fique por dentro das principais novidades sobre coroideremia.
Preencha o formulário abaixo e entraremos em contato em breve.
Solicitar orçamento
Nossa equipe entrará em contato com você em breve.
Fechar
…complete suas informações
Depois
Obrigado por completar suas informações
FECHAR
Depois
Solicite e comece SEU TEST DRIVE
Por favor, preencha o formulário abaixo que entraremos em contato.
Obrigado!
Nosso time comercial logo entrará em contato para finalizar o processo.
FECHAR
Later
Solicite mais informações
Por favor preencha o formulário abaixo e entraremos em contato com você.
Obrigado!
Nossa equipe comercial entrará em contato em breve para finalizar o processo. Nossa equipe comercial entrará em contato em breve para finalizar o processo.