

Pesquisadores do National Eye Institute (NEI), dos Estados Unidos, desenvolveram uma terapia gênica que teve resultados promissores in vitro ao reparar defeitos nas terminações ciliares de fotorreceptores retinianos de pacientes afetados por um tipo de amaurose congênita de Leber (LCA), doença que causa cegueira na primeira infância.
As descobertas não apenas esclarecem a função da proteína NPHP5 no cílio primário, mas também levam a um tratamento potencial para essa condição de cegueira. O trabalho foi divulgado recentemente no periódico Stem Cell Reports.
Em seguida, veja como foi feita a pesquisa e como os resultados podem ajudar na terapia de um dos tipos de LCA.
Amaurose congênita de Leber (LCA)
A amaurose congênita de Leber (LCA) é uma doença genética rara que leva à degeneração da retina pela morte de fotorreceptores.
Defeitos em pelo menos 25 genes diferentes podem causar o problema. Atualmente, existe tratamento de terapia genética apenas para um tipo de LCA.
O tipo de LCA causado por mutações em NPHP5 é relativamente raro. Causa cegueira em todos os pacientes e, em muitos casos, também pode levar à insuficiência renal, uma condição chamada Síndrome de Senior-Løken.
A pesquisa
Usando organoides (fabricados a partir de fibroblastos) que foram transformados em epitélio pigmentar de retina derivados de dois pacientes (também conhecidos como retinas-in-a-dish), os pesquisadores descobriram que um tipo de LCA causado por mutações no gene NPHP5 (também chamado IQCB1) leva a defeitos graves no cílio primário das células fotorreceptoras retinianas.
Os organoides são valiosos porque imitam de perto o genótipo e a apresentação da doença da retina em pacientes reais e fornecem um ambiente de tecido “semelhante ao humano” para testar intervenções terapêuticas, incluindo terapias genéticas.
Como nos pacientes, esse material apresentou defeitos nos fotorreceptores, incluindo a perda da porção do fotorreceptor chamada “segmentos externos”.
Em um olho saudável, acredita-se que a proteína codificada pelo gene NPHP5 fique em uma estrutura semelhante a um portão na base do cílio primário que ajuda a filtrar as proteínas que entram no cílio.
Estudos anteriores em camundongos mostraram que o NPHP5 está envolvido no cílio, mas os pesquisadores ainda não sabem o papel exato no cílio fotorreceptor, nem está claro exatamente como as mutações afetam a função da proteína.
Os resultados
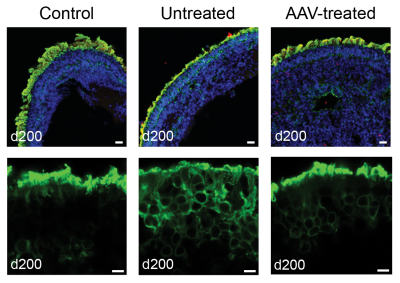
O tratamento de organoides da retina derivados do paciente com AAV-NPHP5 restaura a localização da opsina nos segmentos externos dos fotorreceptores. Acima: retinóides corados para DNA (azul), NPHP5 (vermelho) e opsina (verde). Abaixo: visão aproximada da camada de fotorreceptores organoides corada de verde para a opsina. Adaptado de Kruczek et al, 2022. Crédito da imagem: Anand Swaroop, Ph.D. e Kamil Kruczek, Ph.D.
Além de encontrarem níveis reduzidos da proteína NPHP5 nas células organoides da retina derivadas do paciente, os pesquisadores descobriram níveis menores de outra proteína chamada CEP-290, que interage com o NPHP5 e forma o portão primário do cílio. As mutações no CEP-290 constituem a causa mais comum de LCA.
Outro achado é que os segmentos externos dos fotorreceptores nos organoides da retina estavam completamente ausentes, e a proteína opsina, que deveria ter sido localizada nos segmentos externos, foi encontrada em outro lugar no corpo da célula fotorreceptora.
Quando os pesquisadores introduziram um vetor viral adeno-associado (AAV) contendo uma versão funcional do NPHP5 como veículo de terapia genética, os organoides da retina mostraram uma restauração significativa da proteína opsina concentrada no local apropriado nos segmentos externos.
Os achados também sugerem que o NPHP5 funcional pode ter estabilizado o portão do cílio primário. Dessa forma, as descobertas não apenas apontam para uma função da proteína NPHP5 no cílio primário, mas também levam a caminho potencial para tratamento no futuro dessa condição de cegueira.
Revisado por Paulo Schor, médico oftalmologista, professor livre docente e diretor de inovação da Universidade Federal de São Paulo (Unifesp) e colaborador da Faculdade de Medicina do Hospital Albert Einstein.
Acompanhe o blog da Phelcom e fique por dentro das principais novidades sobre amaurose congênita de Leber (LCA).



